Zebrafish Can Repair Their Brains. Why Can’t We?

Photo: National Institute of Child Health and Human Development
Evolution is usually equated with advancement, but the most useful biological innovations aren’t always found in the “highest” organisms.
Zebrafish, for example, can continually generate new neurons in their brains, which protects these vertebrates from the ravages of neurological disease and injury. Adult humans, on the other hand, can only make a meager number of new neurons, mostly in the hippocampus (the brain’s memory center), and diseases like Alzheimer’s suppress this production even further.
Fortunately, zebrafish are giving up their secrets.
Columbia University researchers have discovered a mechanism that promotes neurogenesis in the zebrafish brain and has the potential to be activated in people.
“It’s possible that by activating neurogenesis in people with Alzheimer’s disease, we could slow the disease’s progression,” says study leader Caghan Kizil, PhD, associate professor of neurological sciences (in neurology and in the Taub Institute).
Growth factor helps zebrafish regenerate their brains
Kizil and his colleagues previously found a molecule in zebrafish brains called nerve growth factor receptor (NGFR) that nudges stem cells to make new neurons.
NGFR increases neuron production even in zebrafish whose brains show signs of Alzheimer’s disease, which impedes neurogenesis.
Mammals have the same regeneration machinery
In the new study, the researchers wanted to determine if the zebrafish trick could also work in mammals with Alzheimer’s disease.
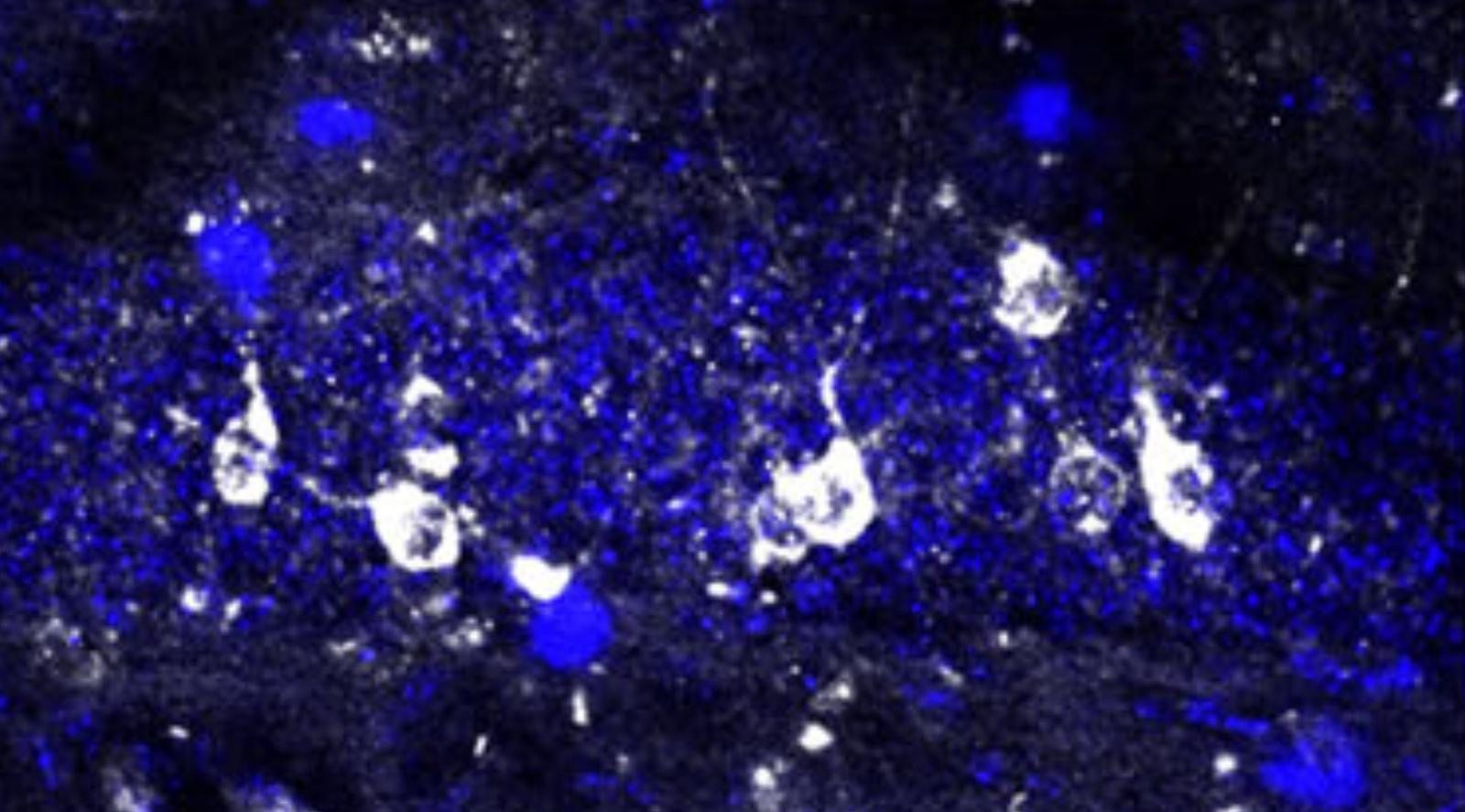
Mouse brains do not normally express NGFR, but when the researchers activated it in mouse models of Alzheimer’s, they found that NGFR stimulated long-term neurogenesis by converting the disease-related physiology of the brain’s astrocytes to a healthy and neurogenic state. Remarkably, NGRF activation also reduced the accumulation of amyloid and tau proteins, two of the hallmarks of Alzheimer’s disease.
Analysis of human brain cells, including those from deceased Alzheimer’s patients, showed that NGFR is expressed in the brain during early human development but not later in life.
How to activate the zebrafish trick in people
Turning on NGFR in relevant cell types could be a therapy option yet it would require gene therapy approaches. Instead, Kizil and his colleagues deciphered how NGFR works to promote neurogenesis to find other synergistic targets that could be modified. They identified two potential drug targets that act downstream to NGFR.

The first target, lipocalin-2, is a protein that is elevated in mouse and human brain cells and must be blocked for neurogenesis to proceed. “Our findings suggest that if we simply block lipocalin-2’s receptor—Slc22a17—we might be able to increase neurogenesis,” says Kizil.
The researchers also identified a second protein called phosphofructokinase that also hampers neurogenesis and is highly expressed in human brain cells. And because phosphofructokinase works independently from lipocalin-2, it might be more effective to go after both targets simultaneously, Kizil says.
What’s next?
In vivo studies and behavioral tests in preclinical Alzheimer’s models are needed to determine if more neurons and less Alzheimer’s pathology would lead to cognitive improvement.
References
More information
The study, “Nerve growth factor receptor (Ngfr) induces neurogenic plasticity by suppressing reactive astroglial Lcn2/Slc22a17 signaling in Alzheimer’s disease,” was published in npj Regenerative Medicine on July 10.
The other contributors: Tohid Siddiqui (German Center for Neurodegenerative Diseases within Helmholtz Association, Dresden, Germany), Mehmet Ilyas Cosacak (German Center for Neurodegenerative Diseases within Helmholtz Association), Stanislava Popova (German Center for Neurodegenerative Diseases within Helmholtz Association and now at Neuron D GmbH, Tatzberg, Dresden, Germany), Prabesh Bhattarai (Columbia), Elanur Yilmaz (Columbia), Annie J. Lee (Columbia), Yuhao Min (Mayo Clinic Florida, Jacksonville, Florida), Xue Wang (Mayo Clinic Florida), Mariet Allen (Mayo Clinic Florida), Özkan İş (Mayo Clinic Florida), Zeynep Tansu Atasavum (German Center for Neurodegenerative Diseases within Helmholtz Association), Natalia Rodriguez-Muela (German Center for Neurodegenerative Diseases within Helmholtz Association), Badri N. Vardarajan (Columbia), Delaney Flaherty (Columbia), Andrew F. Teich (Columbia), Ismael Santa-Maria (Columbia and Universidad Francisco de Vitoria, Edificio E, Pozuelo de Alarcon, Madrid, Spain), Uwe Freudenberg (Leibniz-Institut für Polymerforschung Dresden, Dresden, Germany), Carsten Werner (Leibniz-Institut für Polymerforschung Dresden and Cluster of Excellence Physics of Life, TU Dresden, Dresden, Germany), Giuseppe Tosto (Columbia), Richard Mayeux (Columbia), and Nilüfer Ertekin-Taner (Mayo Clinic Florida).
This work was supported by the German Center for Neurodegenerative Diseases, a Columbia University Schaefer Research Scholar Award, the Thompson Family Foundation Program for Accelerated Medicines Exploration in Alzheimer’s Disease and Related Disorders of the Nervous System, Taub Institute Grants for Emerging Research, the National Institute on Aging (grants U01AG046139 and R01AG061796), and Alzheimer’s Association Zenith Awards.
Caghan Kizil is a scientific advisor and Stanislava Popova is a current employee of Neuron D GmbH, which did not have any influence on or financial relationship to the current study. The remaining authors declare no competing interests.